brownbag-science
Next-Generation Sequencing (NGS)
Just a wee-bit complicated
(Extremely) Generic Steps
- (Sample) –>
- Amplified/Template DNA –>
- Short Reads from Sequencer (FASTQ) –>
- Mapped reads (BAM) –>
- sequence !
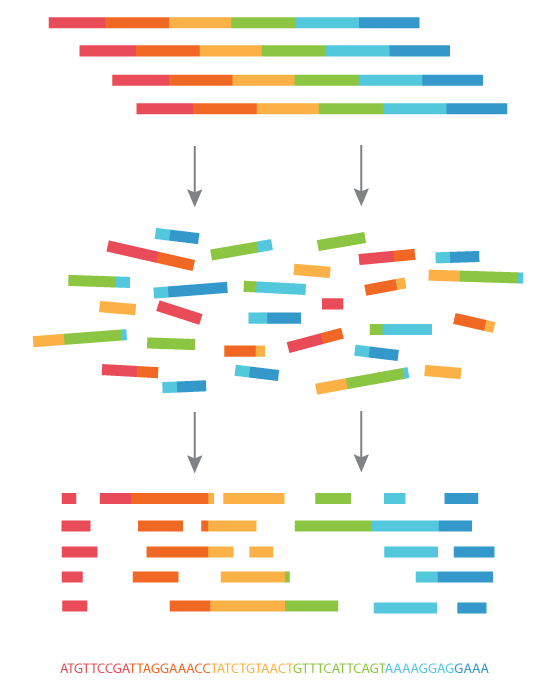
(figure taken from abmgood.com)
Overall Steps - More Detail
- Sample preparation (extract dna from biological sample)
- Library Preparation, Construction (chop up DNA into fragments)
- Sequencing
- raw sequencing reads captured: BCL
- (on sequencer) BCL converted and then converted FASTQ
- FASTQ = FASTA + quality info
- Sequence identifier
- Nucleotide sequence - aka the read
- Phred quality info per base
- FASTQ = FASTA + quality info
- Data Analysis
- Quality Control, including trimming and/or filtering reads ()
- Align reads to reference genome: FASTQ –> SAM/BAM
- Alignment Cleanup: BAM
- Variant Calling (VCF)
- Variant Annotation
- Add information for each variant: symbol, name, transcript, amino acid sequence
- COSMIC, dbSNP/1000 genomes, panel of normals, etc
- Variant Filtering - Biological
- In a clinical setting, usually no matched normal –> Remove unimportant variants
- Remove known germline variants in population; Improving databases (e.g. dbSNP -> 1000 genomes -> 1000 genomes -> SweGen)
- Variant Filtering - Technical
- VAR cutoff, read depth, variant quality score
- Panel of normals
- General Quality Control
- Base quality, GC bias, distinct reads, percent reads mapped, mapping quality, average depth on target region, uniformity, etc
Data Analysis
Steps in a typical next-generation resequencing workflow. De facto standard file formats are given in parentheses.
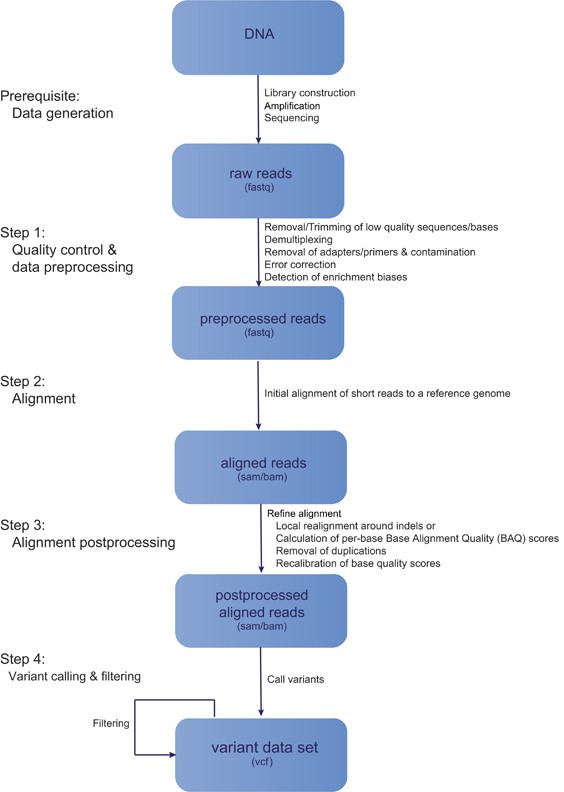
(figure taken from nature.com)
Prev: 05-Medical-Genetics.md
Next: 07-Types-of-NGS.md